
Rare diseases


Scroll down to see updated content for unmet need in rare disease.
Explore our entire portfolio of FDA-approved products in rare diseases
Our current portfolio includes approved treatments for:






Search for other FDA-approved products
ERT, enzyme replacement therapy; FDA, US Food and Drug Administration; SRT, substrate reduction therapy.

-

Our legacy in R&D: Lysosomal storage disorders
Lysosomal storage disorders (LSDs) are a group of rare, inherited disorders characterized by the absence of vital enzymes that break down specific fats and sugars1
LSDs are complex, progressive, difficult to diagnose, and potentially life-threatening
The progression rate, symptom severity, and affected organs differ across disorder type.
There are several reasons it can be difficult for physicians to diagnose LSDs.
![]()
Diagnosis can be difficult primarily because each disorder is rare and symptoms vary among the different disorder types. The physician may confirm a diagnosis once they recognize a pattern of symptoms, but finding that pattern and ruling out other conditions may take years.
![]()
Researchers have identified more than 40 types of LSDs, with Gaucher disease and Fabry disease among the most common forms.
Sanofi’s Gaucher vision centers on 3 core aspects: our heritage, efficacy, and commitment

Gaucher disease is an inherited condition that affects approximately 6,000 people in the United States2
There are 3 forms of Gaucher disease, with type 1 (GD1) affecting 95% of all diagnosed patients in the United States. GD1 is caused by pathogenic variants that result in a deficiency of a functional enzyme called beta-glucocerebrosidase. In the absence of beta-glucocerebrosidase, glucocerebroside (GL-1) accumulates inside cells in various organ systems. In time, this accumulation can lead to the onset of manifestations such as:
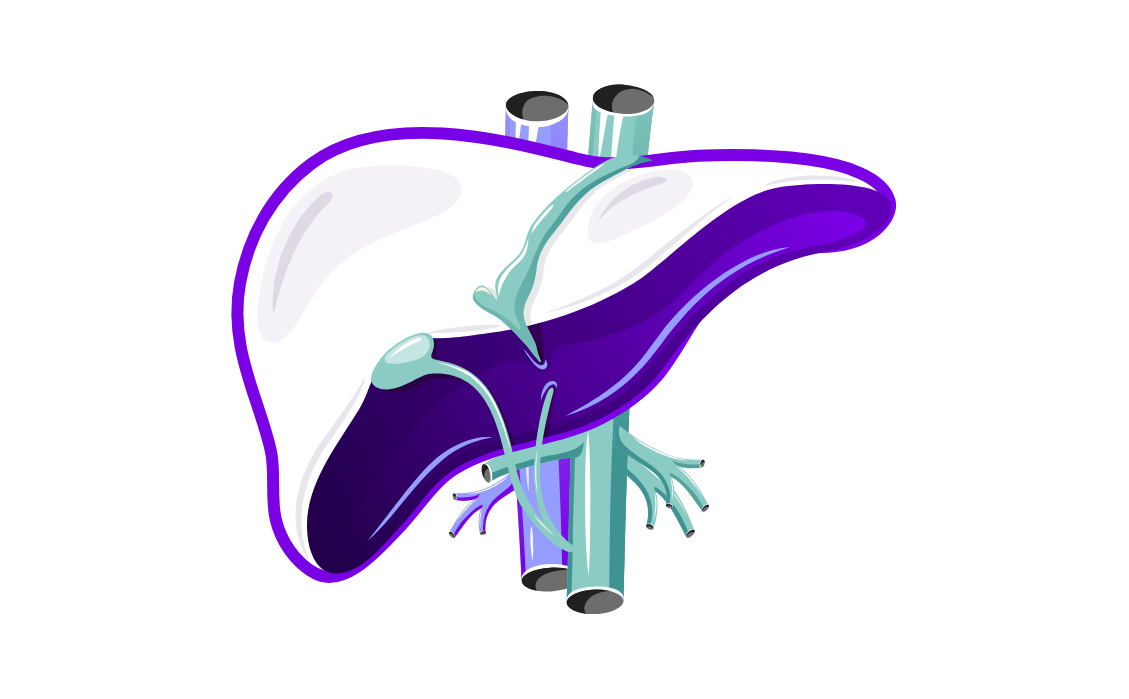
Liver and spleen enlargement
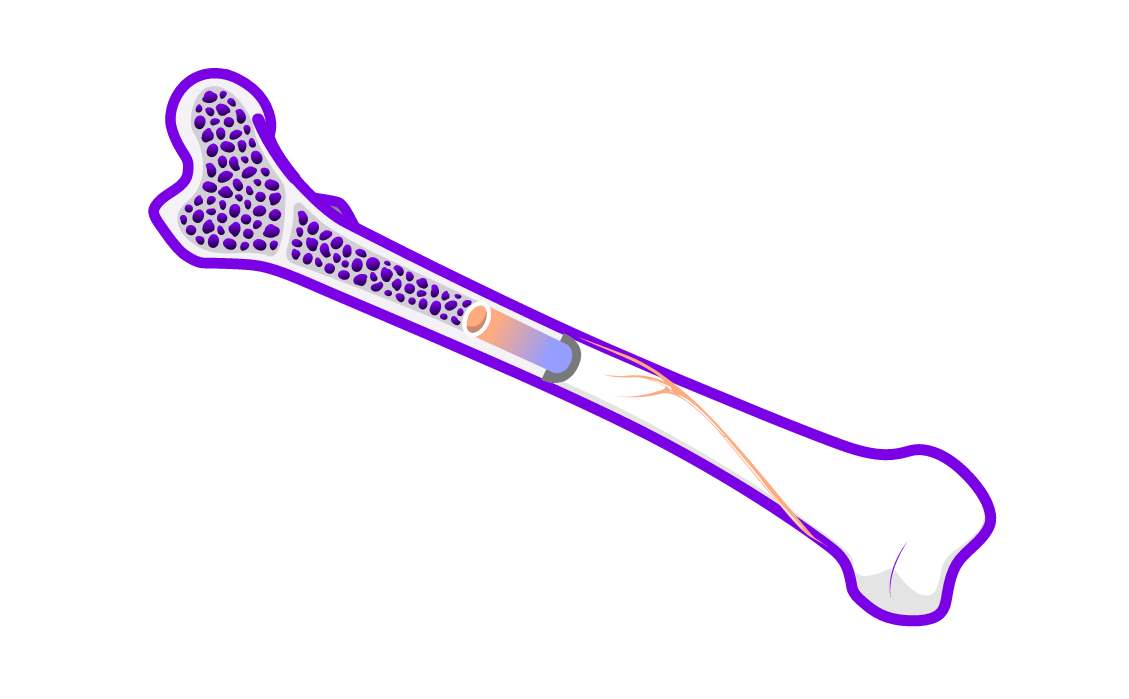
Bone manifestations

Hematologic complications
GD1 varies widely in range and severity of symptoms, age of symptom onset, and rate of progression. Available treatment options work in different ways—either by replacing the missing enzyme or partially inhibiting the production of GL-1. 3
With SRT and ERT, physicians have options that address a broad range of patient needs in
Gaucher disease type 1

Explore our other areas of focus
Immunology and
inflammation

Neurology

Oncology

Rare blood disorders

Explore our science
Review the ongoing pipeline trials across therapeutic areas.

References: 1. Lysosomal storage disease & disorder. National Gaucher Foundation. Accessed May 14, 2025. https://www.gaucherdisease.org/about-gaucher-disease/what-is/lysosomal-storage-disorders/ 2. Gaucher disease. Cleveland Clinic. Updated August 21, 2023. Accessed May 14, 2025. https://my.clevelandclinic.org/health/diseases/16234-gaucher-disease 3. Gaucher disease treatment. National Gaucher Foundation. Accessed May 14, 2025. https://www.gaucherdisease.org/gaucher-diagnosis-treatment/treatment/.
This site is intended for US payers only.
© 2025 Sanofi. All rights reserved.
Gaucher disease popup
Gaucher disease type 3

Click the link below to better understand the spectrum, challenges, and need for a new approach in Gaucher disease type 3 management