
NEXVIAZYME: Clinical evidence
Knowledge of how a treatment impacts the underlying condition is important when evaluating the clinical value that it brings
Mechanism of action
The role of mannose 6-phosphate (M6P) in ERT internalization

NEXVIAZYME is an enzyme replacement therapy (ERT) produced by recombinant DNA technology.1
NEXVIAZYME provides an exogenous source of the deficient GAA enzyme in Pompe disease by entering cells through an M6P-dependent pathway without the need for an enzyme stabilizer.1,2 The ~15-mol M6P per mol enzyme on the surface of NEXVIAZYME binds onto muscle-cell M6P receptors with high affinity, which helps internalize NEXVIAZYME into the cell and facilitate transport to the lysosome.1,3

What is M6P?
M6P is a residue that binds with receptors, mediating the uptake of NEXVIAZYME into muscle cells.1,3,4
NEXVIAZYME: Clinical trials
COMET trial: The first pivotal, head-to-head ERT clinical trial in LOPD1,5

|
NEXVIAZYME TRIAL STUDY DESIGN |
| Study design |
In the PAP (from baseline to week 49), 100 treatment-naive participants with LOPD were randomized (1:1) to receive NEXVIAZYME or alglucosidase alfa. In the ETP (after week 49), participants who received NEXVIAZYME in the PAP continued on treatment (NEXVIAZYME arm) and participants who received alglucosidase alfa in the PAP switched to NEXVIAZYME (switch arm). |
| Select baseline characteristics |
Upright forced vital capacity (FVC) levels ≥32% and ≤85% predicted 6MWT distance between ≥118 m and ≤630 m Median age was 49 years (range from 16 to 78) |
| Primary endpoint | Change in FVC (% predicted) in the upright position from baseline to week 49 |
| Key secondary endpoint | Change in total distance walked in 6 minutes (6MWT) from baseline to week 49 |
*Screening phase (up to 14 days) may be extended up to 8 weeks in prespecified situations. †Randomized at a 1:1 ratio with stratification factors based on baseline FVC, sex, age, and country (Japan or ex-Japan). ‡NEXVIAZYME infusion, safety assessments, and efficacy evaluations.
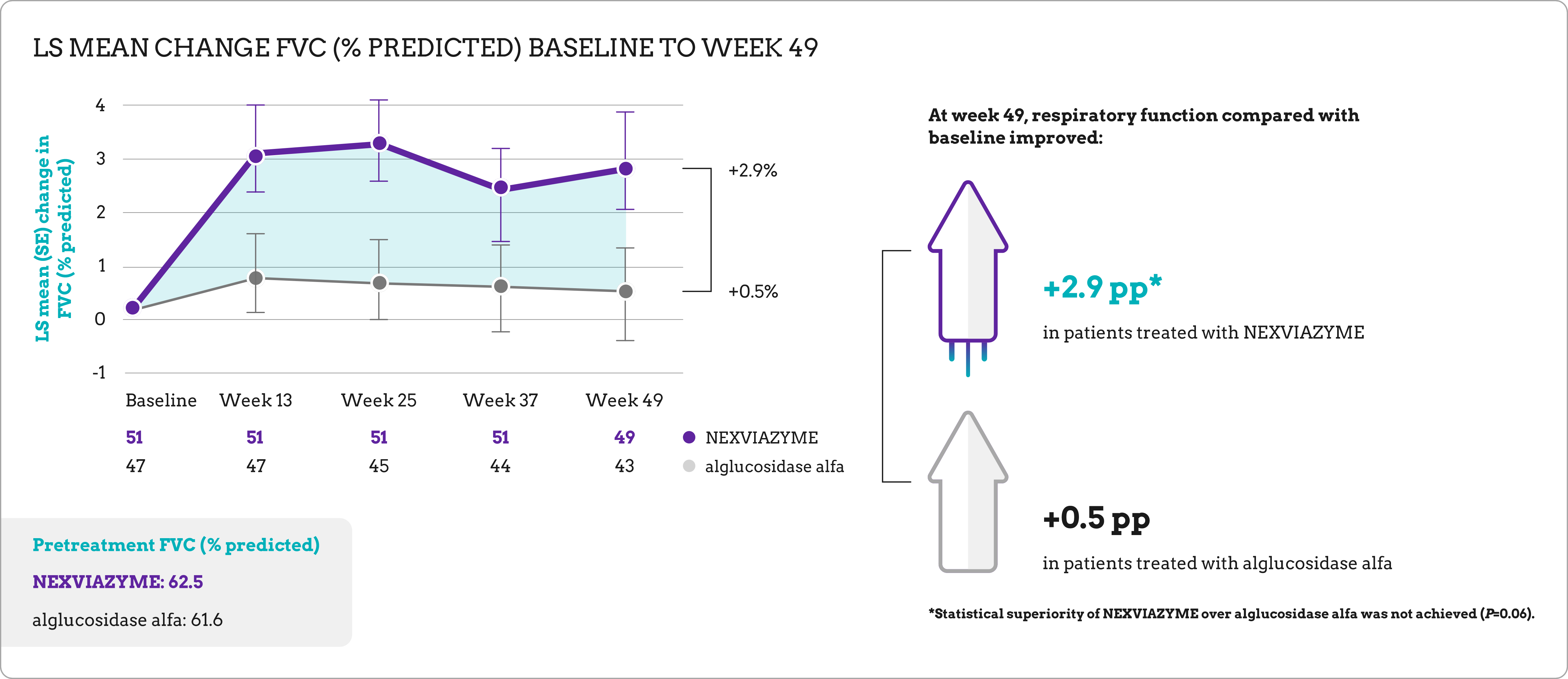
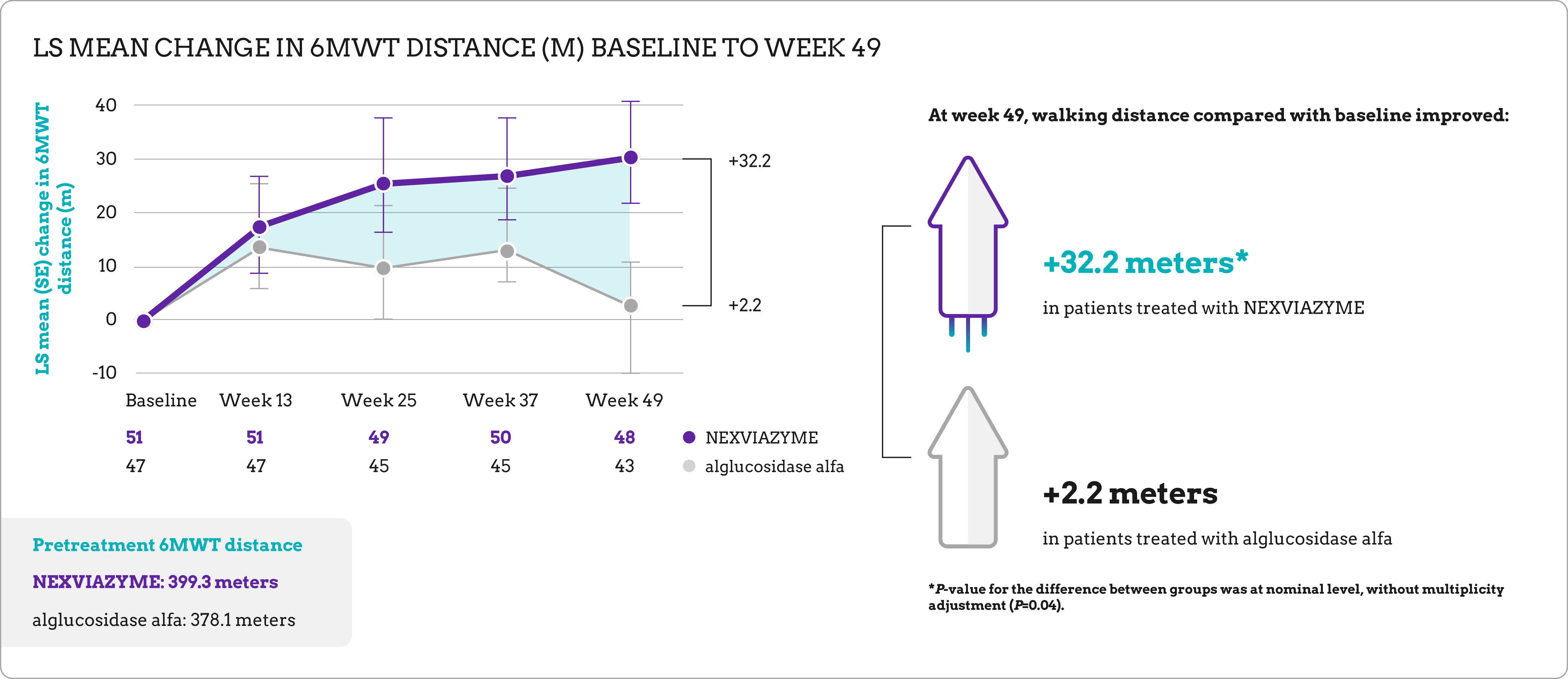
Long-term data for previously treated and switch patients1
Analysis of patients from COMET continued via an open-label extension up to week 145.
The ETP involved patients from the PAP who had previously received either NEXVIAZYME (NEXVIAZYME arm) or alglucosidase alfa (switch arm).
At week 145, patients in both study arms maintained respiratory function and walking ability near baseline.
NEXVIAZYME-Clinical-evidence-popup-1
LS mean change in FVC (% predicted) baseline to week 1451,5


NEXVIAZYME-Clinical-evidence-popup-2
LS mean change in 6MWT (m) baseline to week 1451,5


| RESPIRATORY FUNCTION
|
NEXVIAZYME arm | Switch arm
|
| Baseline mean % predicted FVC | 62.5 (SD=14.4)
|
61.6 (SD=12.4) |
| Mean change % predicted FVC
from baseline at week 145 |
+1.7 (SD=8.6) |
+0.5 (SD=8.3) |
| WALKING ABILITY | NEXVIAZYME arm | Switch arm |
| Baseline mean 6MWT (m) | 399.3 (SD=110.9) | 378.1 (SD=116.2) |
| Mean change from baseline (m)
at week 145 |
+24.9 (SD=68.6) | -4.1 (SD=90.4) |
Safety
Most common adverse reactions reported in COMET1
The pivotal trial was not designed to demonstrate a statistically significant difference in the incidence of adverse reactions in the NEXVIAZYME and the alglucosidase alfa treatment groups.
ADVERSE REACTIONS REPORTED IN AT LEAST 6% OF NEXVIAZYME-TREATED PATIENTS WITH LOPD DURING 49-WEEK PAP
|
ADVERSE REACTION |
NEXVIAZYME (N=51), n (%) |
Alglucosidase alfa (N=49), n (%) |
| Headache | 11 (22%)
|
16 (33%) |
| Fatigue |
9 (18%) |
7 (14%) |
| Diarrhea |
6 (12%) |
8 (16%) |
| Nausea |
6 (12%) |
7 (14%) |
| Arthralgia |
5 (10%) |
8 (16%) |
| Dizziness |
5 (10%) |
4 (8%) |
| Myalgia |
5 (10%) |
7 (14%) |
| Pruritus |
4 (8%) |
4 (8%) |
| Vomiting |
4 (8%) |
3 (6%) |
| Dyspnea |
3 (6%) |
4 (8%) |
| Erythema |
3 (6%) |
3 (6%) |
| Paresthesia |
3 (6%) |
2 (4%) |
| Urticaria |
3 (6%) |
1 (2%) |
Additional safety information1
Infusion-associated reactions (IARs): During the pivotal study, IARs were reported in 25% (13/51) of patients treated with NEXVIAZYME and 33% (16/49) of patients treated with alglucosidase alfa. Mild-to-moderate IARs reported in more than 1 patient with NEXVIAZYME were headache, diarrhea, pruritus, urticaria, and rash. No patients receiving NEXVIAZYME in this study experienced severe IARs.
Serious adverse reactions: Serious adverse reactions were reported in 2% (1/51) of patients treated with NEXVIAZYME.
Pediatric use: The safety and effectiveness of NEXVIAZYME for the treatment of LOPD have been established in patients aged 1 year and older. The safety profile in pediatric patients aged 1 to 12 years was similar to the safety profile in older pediatric and adult patients with LOPD.
Safety and effectiveness have not been established in patients younger than age 1 year. NEXVIAZYME is not approved for the treatment of infantile-onset Pompe disease.
Overall safety profile during ETP5
No new safety concerns were observed during the ETP. Safety data include analysis of both patients treated with NEXVIAZYME and patients who switched from placebo to NEXVIAZYME at the end of the 49-week period.
TREATMENT-EMERGENT ADVERSE EVENTS IN NEXVIAZYME-TREATED PATIENTS WITH LOPD DURING 49-WEEK PAP
|
ADVERSE EVENTS |
NEXVIAZYME arm PAP + ETP (n=51) |
Switch arm ETP* (n=44) |
| Any TEAEs |
50 (98%) |
43 (98%) |
| TEAEs potentially related to treatment |
27 (53%) |
25 (57%) |
| Serious TEAEs |
18 (35%) |
12 (27%) |
| Serious TEAEs potentially related to treatment |
4 (8%) |
2 (5%) |
| Severe TEAEs |
12 (24%) |
10 (23%) |
| TEAEs leading to permanent treatment discontinuation† |
2 (4%) |
3 (7%) |
| TEAEs leading to death |
0 |
1 (2%)‡ |
| Protocol- defined IARs§ |
21 (41%) |
21 (48%) |
Numbers reported are the number (%) of participants with at least 1 treatment-emergent adverse event (TEAE) in each category. The mean (SD) duration of exposure during the ETP at the data cut-off date (March 11, 2022) was 141.19 (45.31) weeks (range, 20.00-223.50 weeks) for the NEXVIAZYME arm and 138.27 (53.55) weeks (range, 7.83-230.02 weeks) for the switch arm.
*Data for the switch arm are for the ETP only while participants were receiving avalglucosidase alfa; this group had received alglucosidase alfa during the PAP. †Five participants discontinued treatment during the ETP by week 145 for 6 adverse events (treatment related: ocular hyperemia and erythema [experienced by the same participant], urticaria, respiratory distress; non–treatment related: acute myocardial infarction, pancreatic adenocarcinoma). ‡Pancreatic adenocarcinoma considered not related to treatment. §Defined as an adverse event that occurred during either the infusion or observation period following the infusion, related or possibly related to the investigational treatment.
Dosing and administration1
Recommended dosing of NEXVIAZYME is every 2 weeks. NEXVIAZYME is for intravenous infusion only.
NEXVIAZYME-Clinical-evidence-popup-3
Administer infusion incrementally, as determined by the patient’s response and comfort1
If there are no signs of IARs, and at the discretion of the healthcare provider, gradually increase the infusion rate every 30 minutes as detailed below.


Recommended dosage for patients with LOPD varies by weight


The recommended starting infusion rate is 1 mg/kg/hr. The approximate infusion duration is between 4 and 7 hours, and is based on dose and patient response.
Prior to NEXVIAZYME administration, healthcare providers should consider pretreating with antihistamines, antipyretics, and/or corticosteroids.
Administration: Sites of care
Based on the recommendations of the treating physician, NEXVIAZYME is expected to be infused at the following locations*:
![]()
Hospital outpatient
![]()
Home infusion
![]()
Freestanding ambulatory care clinics
![]()
Physician offices
![]()
Hospital inpatient
![]()
Federally qualified health centers
*Based on current ERT infusion data.
Administration: Site of care utilization
Patients initiating treatment may start in an inpatient setting before transitioning to outpatient or home infusion.



Connect with us
Schedule a meeting with a member of the Sanofi Market Access team to learn more about Sanofi products.
6MWT, 6-minute walk test; CI, confidence interval; DNA, deoxyribonucleic acid; GAA, acid alpha-glucosidase; IV, intravenous; LOPD, late-onset Pompe disease; LS, least squares; m, meter; qow, every other week; SD, standard deviation; SE, standard error.
References: 1. NEXVIAZYME (avalglucosidase alfa). Prescribing information. Genzyme Corporation; 2021. 2. Reuser AJ, Kroos MA, Ponne NJ, et al. Uptake and stability of human and bovine acid α- glucosidase in cultured fibroblasts and skeletal muscle cells from glycogenosis type II patients. Exp Cell Res. 1984;155(1):178-189. doi:10.1016/0014-4827(84)90779-1 3. Pena LDM, Barohn RJ, Byrne BJ, et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naïve and alglucosidase alfa- treated patients with late-onset Pompe disease: a phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul Disord. 2019;29(3):167-186. doi:10.1016/j.nmd.2018.12.004 4. Zhu Y, Jiang JL, Gumlaw NK, et al. Glycoengineered acid α-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther. 2009;17(6):954-963. doi:10.1038/mt.2009.37 5. Straub V, Kishnani PS, Diaz-Manera J, et al. Efficacy and safety of avalglucosidase alfa in participants with late-onset Pompe disease after 145 weeks of treatment during the COMET trial. Poster presented at: Muscular Dystrophy Association Clinical & Scientific Conference; March 19-22, 2023; Dallas, TX. Poster 143. 6. Data on file. Sanofi. 2022.
INDICATION
NEXVIAZYME (avalglucosidase alfa-ngpt) is indicated for the treatment of patients 1 year of age and older with late- onset Pompe disease [lysosomal acid alpha-glucosidase (GAA) deficiency].
Please see full Prescribing Information for complete details, including Boxed WARNING.
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions Including Anaphylaxis: See Boxed WARNING. Prior to NEXVIAZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids. The risks and benefits of readministering NEXVIAZYME following severe hypersensitivity reaction (including anaphylaxis) should be considered. If a mild or moderate hypersensitivity reaction occurs, the infusion rate may be slowed or temporarily stopped.
Infusion-Associated Reactions: See Boxed WARNING. IARs may still occur in patients after receiving pretreatment. If mild or moderate IARs occur regardless of pretreatment, decreasing the infusion rate or temporarily stopping the infusion may ameliorate the symptoms.
Risk of Acute Cardiorespiratory Failure in Susceptible Patients: See Boxed WARNING.
ADVERSE REACTIONS
The most common adverse reactions (>5%) were headache, fatigue, diarrhea, nausea, arthralgia, dizziness, myalgia, pruritus, vomiting, dyspnea, erythema, paresthesia and urticaria.
INDICATION
NEXVIAZYME (avalglucosidase alfa-ngpt) is indicated for the treatment of patients 1 year of age and older with late- onset Pompe disease [lysosomal acid alpha-glucosidase (GAA) deficiency].
Please see full Prescribing Information for complete details, including Boxed WARNING.
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions Including Anaphylaxis: See Boxed WARNING. Prior to NEXVIAZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids. The risks and benefits of readministering NEXVIAZYME following severe hypersensitivity reaction (including anaphylaxis) should be considered. If a mild or moderate hypersensitivity reaction occurs, the infusion rate may be slowed or temporarily stopped.
Infusion-Associated Reactions: See Boxed WARNING. IARs may still occur in patients after receiving pretreatment. If mild or moderate IARs occur regardless of pretreatment, decreasing the infusion rate or temporarily stopping the infusion may ameliorate the symptoms.
Risk of Acute Cardiorespiratory Failure in Susceptible Patients: See Boxed WARNING.
ADVERSE REACTIONS
The most common adverse reactions (>5%) were headache, fatigue, diarrhea, nausea, arthralgia, dizziness, myalgia, pruritus, vomiting, dyspnea, erythema, paresthesia and urticaria.
INDICATION
NEXVIAZYME (avalglucosidase alfa-ngpt) is indicated for the treatment of patients 1 year of age and older with late- onset Pompe disease [lysosomal acid alpha-glucosidase (GAA) deficiency].
Please see full Prescribing Information for complete details, including Boxed WARNING.
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions Including Anaphylaxis: See Boxed WARNING. Prior to NEXVIAZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids. The risks and benefits of readministering NEXVIAZYME following severe hypersensitivity reaction (including anaphylaxis) should be considered. If a mild or moderate hypersensitivity reaction occurs, the infusion rate may be slowed or temporarily stopped.
Infusion-Associated Reactions: See Boxed WARNING. IARs may still occur in patients after receiving pretreatment. If mild or moderate IARs occur regardless of pretreatment, decreasing the infusion rate or temporarily stopping the infusion may ameliorate the symptoms.
Risk of Acute Cardiorespiratory Failure in Susceptible Patients: See Boxed WARNING.
ADVERSE REACTIONS
The most common adverse reactions (>5%) were headache, fatigue, diarrhea, nausea, arthralgia, dizziness, myalgia, pruritus, vomiting, dyspnea, erythema, paresthesia and urticaria.
This site is intended for US payers only.
© Sanofi. All rights reserved.
NEXVIAZYME is owned by Genzyme Corporation. NEXVIAZYME and Sanofi are registered trademarks of Sanofi or an affiliate.
CareConnect Personalized Support Services is a trademark of Sanofi or an affiliate.
Leave site modal

You are about to leave SanofiMarketAccess website.
Sanofi does not review or control the content of non-Sanofi websites.